Software
![]()
![]()
Software
GROMACS is available to users at HPC2N under the condition that published work include citation of the program. GROMACS is Free Software, available under the GNU General Public License.
GROMACS (GROningen MAchine for Chemical Simulations) is a versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles.
GROMACS was first developed in Herman Berendsens group, department of Biophysical Chemistry of Groningen University. It is a team effort, with contributions from several current and former developers all over world.
GROMACS is a versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles.
It is primarily designed for biochemical molecules like proteins, lipids and nucleic acids that have a lot of complicated bonded interactions, but since GROMACS is extremely fast at calculating the nonbonded interactions (that usually dominate simulations) many groups are also using it for research on non-biological systems, e.g. polymers.
GROMACS supports all the usual algorithms you expect from a modern molecular dynamics implementation.
On HPC2N we have GROMACS available as a module on Kebnekaise.
On Kebnekaise, some of the versions of GROMACS have suffixes:
Newer versions (2016.2 and forward) are built with both, as default.
To use the gromacs module, add it to your environment. Use:
module spider gromacs
or
ml spider gromacs
to see which versions are available, as well as how to load the module and the needed prerequisites. There are several versions.
Loading the module should set all the needed environmental variables as well as the path.
Note that while the case does not matter when you use "ml spider", it is necessary to match the case when loading the modules.
You can read more about loading modules on our Accessing software with Lmod page and our Using modules (Lmod) page.
In order to access the GPU aware version of Gromacs, you need to load one of the versions compiled with CUDA, and then load the prerequisites given that includes CUDA.
$ ml spider GROMACS/2016.4
---------------------------------------------------------------------------------------
GROMACS: GROMACS/2016.4
---------------------------------------------------------------------------------------
Description:
GROMACS is a versatile package to perform molecular dynamics, i.e. simulate the
Newtonian equations of motion for systems with hundreds to millions of
particles. - Homepage: http://www.gromacs.org
You will need to load all module(s) on any one of the lines below before the "GROMACS/2016.4" module is available to load.
GCC/5.4.0-2.26 CUDA/8.0.61_375.26 impi/2017.3.196
GCC/6.4.0-2.27 impi/2017.3.196
Help:
GROMACS is a versatile package to perform molecular dynamics,
i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles. - Homepage: http://www.gromacs.orgYou then need to load
ml GCC/5.4.0-2.26 CUDA/8.0.61_375.26 impi/2017.3.196 ml GROMACS/2016.4
There are some differences between how you run Gromacs versions 4.x and newer versions. The focus in this documentation is on newer versions (mainly version 2016.x). You can find manuals for older versions here: ftp://ftp.gromacs.org/pub/manual/
When you have loaded Gromacs and its prerequisites, you can find the executables etc. under the directory pointed to by the environment variable $EBROOTGROMACS.
You can get some information about the particular version of Gromacs, do
gmx -version
To run Gromacs, you first need to prepare various input files (.gro and .pdb for molecular structure, .top for topology and the main parameters file, .mdp).
The following steps are needed to do your setup (adapted from the Gromacs homepage's Getting started guide):
1) The molecular topology file is generated by the program
gmx pdb2gmx
gmx pdb2gmx translates a pdb structure file of any peptide or protein to a molecular topology file. This topology file contains a complete description of all the interactions in your peptide or protein.
2) When gmx pdb2gmx is executed to generate a molecular topology, it also translates the structure file (pdb file) to a GROMOS structure file (gro file). The main difference between a pdb file and a gromos file is their format and that a gro file can also hold velocities. However, if you do not need the velocities, you can also use a pdb file in all programs.
3) To generate a box of solvent molecules around the peptide, the program
gmx solvate
is used. First the program
gmx editconf
should be used to define a box of appropriate size around the molecule. gmx solvate solvates a solute molecule (the peptide) into any solvent. The output of gmx solvate is a gromos structure file of the peptide solvated. gmx solvate also changes the molecular topology file (generated by gmx pdb2gmx) to add solvent to the topology.
4) The Molecular Dynamics Parameter (mdp) file contains all information about the Molecular Dynamics simulation itself e.g. time-step, number of steps, temperature, pressure etc. The easiest way of handling such a file is by adapting a sample mdp file. A sample mdp file is available from the Gromacs homepage.
5) The next step is to combine the molecular structure (gro file), topology (top file) MD-parameters (mdp file) and (optionally) the index file (ndx) to generate a run input file (tpr extension). This file contains all information needed to start a simulation with GROMACS. The
gmx gromppprogram processes all input files and generates the run input tpr file.
6) Once the run input file is available, we can start the simulation. The program which starts the simulation is called
gmx mdrunor
gmx_mpi mdrun
The only input file of gmx mdrun that you usually need in order to start a run is the run input file (tpr file). The typical output files of gmx mdrun are the trajectory file (trr file), a logfile (log file), and perhaps a checkpoint file (cpt file).
Below is some examples on how to run Gromacs, gmx mdrun or gmx_mpi mdrun, jobs on Kebnekaise. Always note the number of cores per node (28 for a regular Broadwell node), and the amount of memory per node.
Note that you must run from the parallel file system.
When using gmx mdrun (and gmx_mpi mdrun) it is important to specify the -ntomp option. If not gmx(_mpi) mdrun will try to use all the cores on the node by adding multiple OpenMP threads to each (MPI) task. If the batch job does not have the whole node allocated (using --exclusive, --ntasks-per-node=28 for a standard Kebnekaise node, or other means) this will result in overallocation of the cores resulting in severely reduced performance. So, all examples below explicitly use -ntomp to avoid that situation.
#!/bin/bash
# Change to your actual project id number (of the form: hpc2nXXXX-YYY, SNICXXX-YY-ZZ, or NAISSXXXX-YY-ZZ)
#SBATCH -A hpc2nXXXX-YYY
# Asking for 30 hours walltime
#SBATCH -t 30:00:00
# Use 14 tasks
#SBATCH -n 14
# Use 2 threads per task
#SBATCH -c 2
# It is always best to do a ml purge before loading modules in a submit file
ml purge > /dev/null 2>&1
# Load the module for GROMACS and its prerequisites.
# This is for GROMACS/2016.4 with no GPU support
ml GCC/6.4.0-2.28 impi/2017.3.196
ml GROMACS/2016.4
# Automatic selection of single or multi node based GROMACS
if [ $SLURM_JOB_NUM_NODES -gt 1 ]; then
GMX="gmx_mpi"
MPIRUN="mpirun"
ntmpi=""
else
GMX="gmx"
MPIRUN=""
ntmpi="-ntmpi $SLURM_NTASKS"
fi
# Automatic selection of ntomp argument based on "-c" argument to sbatch
if [ -n "$SLURM_CPUS_PER_TASK" ]; then
ntomp="$SLURM_CPUS_PER_TASK"
else
ntomp="1"
fi
# Make sure to set OMP_NUM_THREADS equal to the value used for ntomp
# to avoid complaints from GROMACS
export OMP_NUM_THREADS=$ntomp
$MPIRUN $GMX mdrun $ntmpi -ntomp $ntomp -deffnm md_0
#!/bin/bash
# Change to your actual project id number (of the form: hpc2nXXXX-YYY, SNICXXX-YY-ZZ, or NAISSXXXX-YY-ZZ)
#SBATCH -A hpc2nXXXX-YYY
# Name of the job
#SBATCH -J Gromacs-gpu-job
# Asking for one hour of walltime
#SBATCH -t 01:00:00
# Use for 4 tasks
#SBATCH -n 4
# Usefor 7 threads per task
#SBATCH -c 7
# Remember the total number of cores = 28 (kebnekaise) so that
# n x c = 28 (running on a single node, or multiples of 28 for multi node runs)
# Asking for 2 K80 GPU cards per node
#SBATCH --gres=gpu:k80:2
# It is always best to do a ml purge before loading modules in a submit file
ml purge > /dev/null 2>&1
# Load the module for GROMACS and its prerequisites.
# This is for GROMACS/2016.4 with GPU support
ml GCC/5.4.0-2.26 CUDA/8.0.61_375.26 impi/2017.3.196
ml GROMACS/2016.4
# Automatic selection of single or multi node based GROMACS
if [ $SLURM_JOB_NUM_NODES -gt 1 ]; then
GMX="gmx_mpi"
MPIRUN="mpirun"
ntmpi=""
else
GMX="gmx"
MPIRUN=""
ntmpi="-ntmpi $SLURM_NTASKS"
fi
# Automatic selection of ntomp argument based on "-c" argument to sbatch
if [ -n "$SLURM_CPUS_PER_TASK" ]; then
ntomp="$SLURM_CPUS_PER_TASK"
else
ntomp="1"
fi
# Make sure to set OMP_NUM_THREADS equal to the value used for ntomp
# to avoid complaints from GROMACS
export OMP_NUM_THREADS=$ntomp
$MPIRUN $GMX mdrun $ntmpi -ntomp $ntomp -deffnm md_0
Starting from version 2018, other tasks besides PME long-range part can be offloaded to GPUs
#!/bin/bash # Change to your actual local/SNIC/NAISS id project number (of the form: hpc2nXXXX-YYY, SNICXXX-YY-ZZ, or NAISSXXXX-YY-ZZ) #SBATCH -A hpc2nXXXX-YYY #SBATCH -t 00:05:00 #SBATCH -N 1 # Use for 4 tasks #SBATCH -n 4 # Usefor 7 threads per task #SBATCH -c 7 # Remember the total number of cores = 28 (kebnekaise) so that # n x c = 28 (running on a single node, or multiples of 28 for multi node runs) # Asking for 2 K80 GPU cards per node #SBATCH --gres=gpu:k80:2 # It is always best to do a ml purge before loading modules in a submit file ml purge > /dev/null 2>&1 ml GCC/7.3.0-2.30 CUDA/9.2.88 OpenMPI/3.1.1 ml GROMACS/2019 # Automatic selection of single or multi node based GROMACS if [ $SLURM_JOB_NUM_NODES -gt 1 ]; then GMX="gmx_mpi" MPIRUN="mpirun" ntmpi="" else GMX="gmx" MPIRUN="" ntmpi="-ntmpi $SLURM_NTASKS" fi # Automatic selection of ntomp argument based on "-c" argument to sbatch if [ -n "$SLURM_CPUS_PER_TASK" ]; then ntomp="$SLURM_CPUS_PER_TASK" else ntomp="1" fi # Make sure to set OMP_NUM_THREADS equal to the value used for ntomp # to avoid complaints from GROMACS export OMP_NUM_THREADS=$ntomp $MPIRUN $GMX mdrun -gputasks 0123 -nb gpu -pme gpu -npme 1 $ntmpi -ntomp $ntomp -dlb yes -v -deffnm script_name
In this example, four tasks (3 related to PP interactions and 1 related to the long-range PME part) will be distributed on the 4 GPU engines. Notice that the PME long-range part should be handled by only 1 rank (npme 1). More information about the tasks that can be offloaded can be found in the GROMACS Documentation.
#!/bin/bash
# Change to your actual local/SNIC/NAISS id project number (of the form: hpc2nXXXX-YYY, SNICXXX-YY-ZZ, or NAISSXXXX-YY-ZZ)
#SBATCH -A hpc2nXXXX-YYY
# Name of the job
#SBATCH -J Gromacs-knl-job
# Asking for one hour of walltime
#SBATCH -t 01:00:00
# Use 64 tasks
#SBATCH -n 64
# Request 4 hardware threads per core
#SBATCH --threads-per-core=4
# Ask for core and memory layout
#SBATCH --constraint=hemi,cache
# Ask for knl queue
#SBATCH -p knl
# It is always best to do a ml purge before loading modules in a submit file
ml purge > /dev/null 2>&1
# Load the module for GROMACS and its prerequisites.
# This is for GROMACS/2016.4 on the KNLs
ml icc/2017.4.196-GCC-6.4.0-2.28 ifort/2017.4.196 impi/2017.3.196
ml GROMACS/2016.4
# Automatic selection of single or multi node based GROMACS
if [ $SLURM_JOB_NUM_NODES -gt 1 ]; then
GMX="gmx_mpi"
MPIRUN="mpirun"
ntmpi=""
else
GMX="gmx"
MPIRUN=""
ntmpi="-ntmpi $SLURM_NTASKS"
fi
# Automatic selection of ntomp argument based on "-c" argument to sbatch.
# GROMACS on our KNLs usually runs fastest using only 2 hardware threads per core
# The recommendation is therefore to NOT specify any -c argument to sbatch
# But just in case it is done this code handles that too
if [ -n "$SLURM_CPUS_PER_TASK" ]; then
ntomp="-ntomp $SLURM_CPUS_PER_TASK"
OMP_NUM_THREADS=$SLURM_CPUS_PER_TASK
else
# Allocate two hardware threads per core and pin them.
ntomp="-pin on -pinoffset 0 -pinstride 2 -ntomp 2"
OMP_NUM_THREADS=2
fi
export OMP_NUM_THREADS
$MPIRUN $GMX mdrun $ntmpi $ntomp -deffnm md_0
Submit the script with
sbatch <scriptname>
In the case of KNL nodes we obtained the best performance by using Hemisphere cluster mode (hemi keyword above).
Using 68 cores, which is the actual number of cores on the KNLs, will result in gromacs complaining with
Fatal error: The number of ranks you selected (68) contains a large prime factor 17
So if one wants to use all cores on the node, one has to explicitly set -npme to a good value. A bad npme setting will cause performance problems. Use "gmx tune_pme" to find a good value.
More information about batchscripts here.
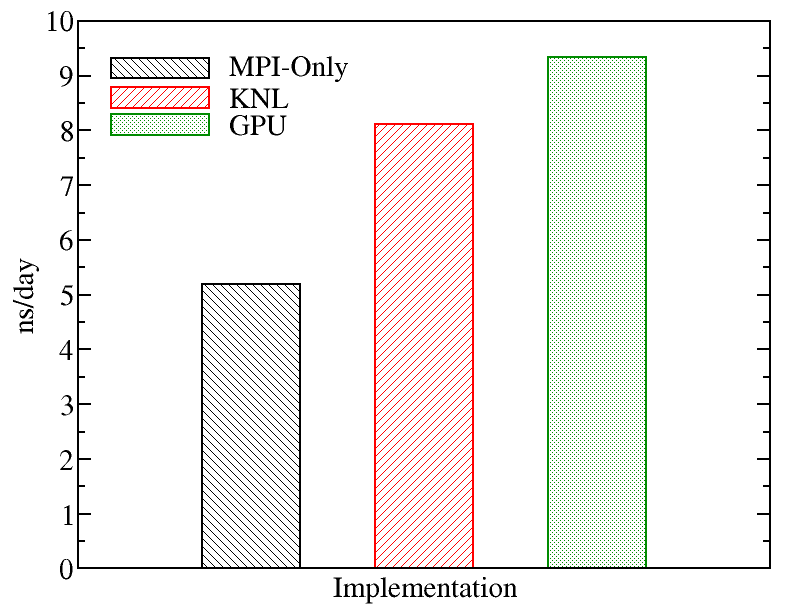
A comparison of runs on the various types of nodes on Kebnekaise is displayed below. We evaluated the performance of different GROMACS implementations including MPI-Only (with 28 cores), KNL (with 272 MPI processes), and GPU (with 4 MPI processes and 7 OpenMP threads). The figure below shows the best performance of GROMACS on a single node obtained by changing the values of input parameters (MPI number of processes and OpenMP number of threads). The benchmark case consisted of 158944 particles, using 1 fs. for time step and a cutoff of 1.2 nm. for real space electrostatics calculations. Particle mesh Ewald was used to solve long-range electrostatic interactions.
Documentation is available on the Gromacs documentation page and the Gromacs Online Reference page.