Software
![]()
![]()
Software
AMBER (Assisted Model Building with Energy Refinement) is freely available to users at HPC2N. You must acknowledge use of Amber in any reports or publications of results obtained with the software.
AMBER is a molecular dynamics platform integrated by independent sub-programs that perform a variety of tasks ranging from integration of Newton's equations to analysis of resulting trajectories. Originally, it simulated the Amber force fields but nowadays it can be used with other common force fields. The AMBER project is currently developed by different research groups including, David Case at Rutgers University, Thomas E. Cheatham III, at the University of Utah, Thomas A. Darden at NIEHS, Kenneth Merz at Michigan State University, Carlos Simmerling at SUNY-Stony Brook University, Ray Luo at UC Irvine, and Junmei Wang at Encysive Pharmaceuticals.
The AMBER software suite provides a set of programs for applying the AMBER forcefields to simulations of biomolecules. It is written in Fortran 90 and C with support for most major Unix-like systems and compilers. Development is conducted by a loose association of mostly academic labs. New versions are generally released in the spring of even numbered years.
The software is available under a site-license agreement. It is available on Kebnekaise.
On HPC2N we have AMBER available as a module. To see which versions are available use:
module spider amber
Read the page about modules, to see how to load the required module.
Note: that Amber needs both C and Fortran libraries, so despite what it says on the output from 'module spider', you need to load both the icc and ifort modules it lists.
If you need to use xleap, or otherwise run applications which opens a display, you must login with
ssh -X
Note: There is rarely any benefit from running multi-gpu jobs on the NVIDIA Volta cards. Here is a quote from the Amber developers:
"With the latest Volta lines it's not possible to get better performance than the single GPU case unless you have some really specialized interconnects and
like eight of them--a corner case that's not worth replicating. Just stick to single GPU runs. The multi-GPU stuff remains in the code because we
will not break functionality that was added years ago, and we may return to the multi-GPU world in time, but for now the benefit of running multiple
GPUs on the Pascal architectures is marginal and Volta is nonexistent."
Prepare the job on the login node (nab, xleap, ...) but do NOT run anything longer/heavier there. You should submit a job script to run sander and pmemd.
There are several tutorials at http://ambermd.org/tutorials/, where you can see how to prepare and run a job with AMBER.
Then, use a batch script similar to one of these, when you submit your job:
The following examples uses "LOAD-THE-MODULE" as a placeholder for the respective module load commands, which can be found with
ml spider amber
For starting MPI enabled programs one should use "srun".
Uses sander.MPI, 8 tasks (cores). It references a groupfile and is derived from the Amber Tutorial A7, found here.
#!/bin/bash #SBATCH -A <Your-Project-Here> #SBATCH -n 8 #SBATCH --time=01:00:00 LOAD-THE-MODULE srun sander.MPI -ng 8 -groupfile equilibrate.groupfile
Uses pmemd.MPI, 96 tasks (cores), 48 per node. It's derived from the Amber Tutorial 17, found here.
#!/bin/bash #SBATCH -A <Your-Project-Here> #SBATCH -n 96 #SBATCH --ntasks-per-node=48 #SBATCH --time=01:00:00 LOAD-THE_MODULE srun pmemd.MPI -O -i 02_heat.in -o 02_heat.out -p ala_tri.prmtop -c 01_min.rst -r 02_heat.rst -x 02_heat.nc
It's useful to note that a groupfile simply specifies file input and output details and is used for convenience. Both sander.MPI and pmemd.MPI can be used with or without a groupfile. If a groupfile is not used, then proper input and output files need to be specified, like in the second example.
Note: that Amber prefers the number of cores to be a power of 2, i.e., 2, 4, 8, 16, 32, 64, etc.
#!/bin/bash #SBATCH -J Amber #SBATCH -A <Your-Project-Here> #SBATCH -n 1 #SBATCH --gres=gpu:v100:1 #SBATCH --time=1:00:00 LOAD-THE-MODULE pmemd.cuda -O -i mdinfile -o mdoutfile -c inpcrdfile -p prmtopfile -r restrtfile
The job is submitted with
sbatch <submitscript.sh>
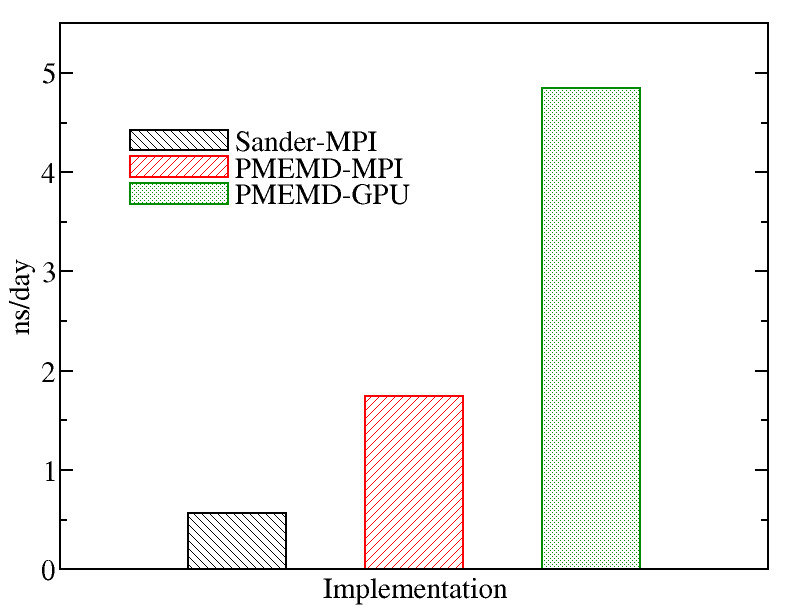
A comparison of runs on the various types of nodes on Kebnekaise is displayed below. We evaluated the performance of different AMBER implementations including Sander-MPI (with 28 cores), PMEMD-MPI (with 28 cores), and PMEMD-GPU (with 1 MPI processes and 1 GPU card). The figure below shows the best performance of AMBER. The benchmark case consisted of 158944 particles, using 1 fs. for time step and a cutoff of 1.2 nm. for real space electrostatics calculations. Particle mesh Ewald was used to solve long-range electrostatic interactions. As a comparison, we used the same test case for Gromacs (see Gromacs page).
More information can be found on the official website.